
Research
Dynamics of water on zirconia and yttria-stabilized zirconia surfaces
This project is in collaboration with the Hou’s group, in which they are investigating ZrO2 surfaces and yttria-stabilized zirconia (YSZ) surfaces on the (100), (110), and (111) crystallographic orientations in contact with an aqueous solution. We use density functional theory (DFT) calculations to obtain energetic and structural information about these systems. Additionally, to provide information on the system’s dynamics and to include temperature effects we use ab initio molecular dynamics (AIMD). Our AIMD simulations are performed at different temperatures as well as with different water surface coverages to determine the effect of chemisorbed water on the stability of the different surfaces.
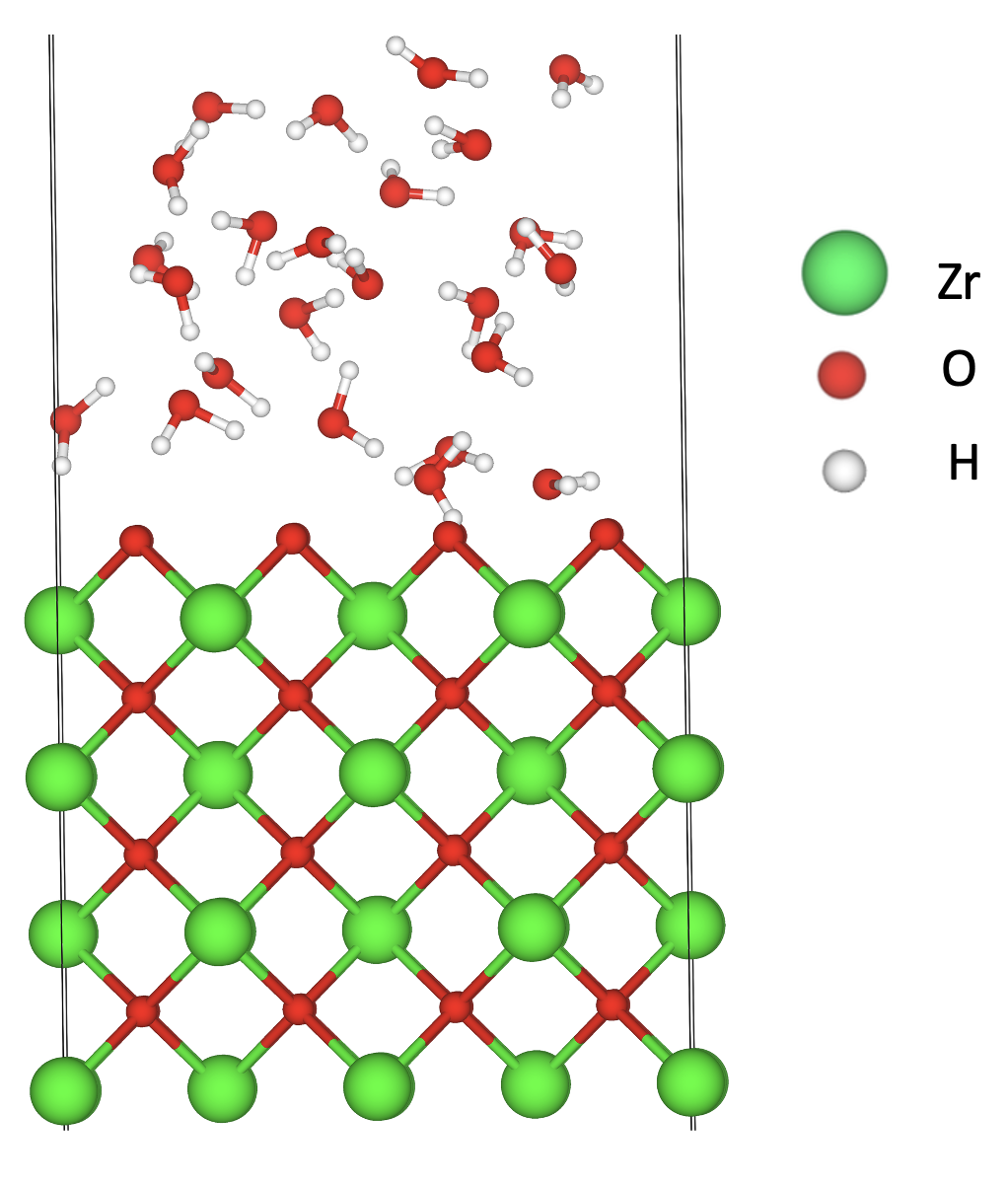
Molecular representation of water molecules physisorption on ZrO2(110) surface.
Knotted protein unfolding
Knotted proteins have been identified in approximately 1% of the protein data bank entries. Knotted proteins are generally topologically more complex than unknotted proteins of similar polypeptide sequence lengths. Additionally, they have a diverse topology with different levels of complexity. Four distinct knot types have been detected in proteins: trefoil, 31, figure-of-eight, 41, three-twist, 52, and stevedore, 61, knots. In this project, we seek to better understand how the presence of a molecular knot affects a protein’s unfolding mechanism compared to a similar unknotted protein. To accomplish this we are studying the free energy surface (FES) of the unfolding/unknotting as a function of the fraction of native contacts. To overcome the high-energy barriers along the free energy surface (FES) of these proteins we use metadynamics.
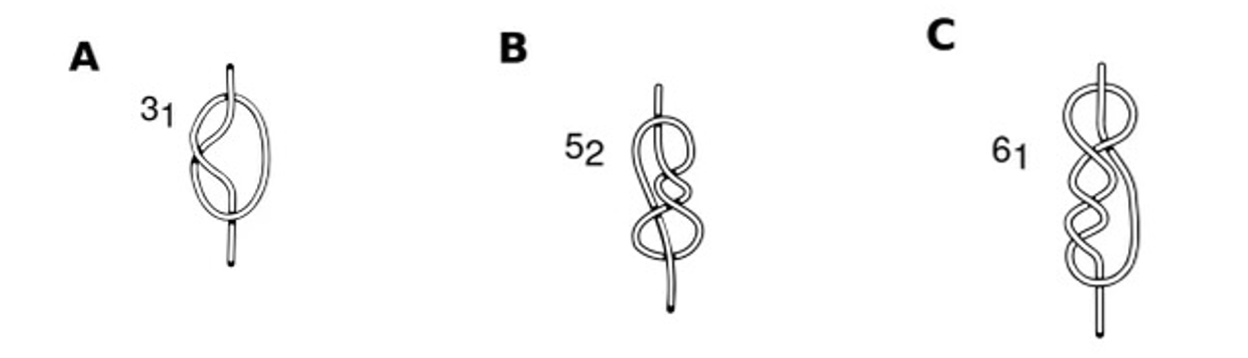
Schematic view of (A) 31 (B) 52 and (C) 61, knots. Figure adapted from Fonseka, Javidi, Oliveira et al. J. Phys. Chem. B 2021, 125, 27, 7335